
产品搜索
结构搜索
全站搜索
在线客服
-
诊断医药客服
当前位置: 行业资讯
无保护氨基酸合成多肽领域的突破性进展
本文转自X-MOL资讯
多肽是由氨基酸按照特定的顺序通过酰胺键(肽键)连接起来的具有重要生物活性的分子,在生命科学、化学、材料和医学等领域发挥着重要作用。由于其靶向选择性高、毒副作用小、体内残留低等优点,受到了药物研发领域的青睐。多肽药物在过去三十年获得了快速发展,目前已有80多种肽药物获得批准,其中不乏有司美格鲁肽(2023年销售额212亿美元)这样的重磅炸弹级别的品种。多肽主要是通过生物合成与化学合成两个途径获得的。除少数多肽药物通过生物途径合成外,大部分多肽药物由于含有化学修饰或非天然氨基酸,难以通过生物方法来制备,只能通过半合成或化学全合成来实现。多肽化学合成迄今已走过一百多年的历程,当前多肽合成主要是通过曾获1984年诺贝尔化学奖的固相多肽合成技术来实现,基于此技术开发的固相多肽合成仪已经实现了多肽合成的自动化。传统多肽合成无论是液相合成还是固相合成都是利用氨基预先保护的α-氨基酸为砌块,经过活化、氨解和去保护这三步实现肽链沿C端向N端的(C→N)增长。整个多肽合成过程就是反复重复 “保护-活化-氨解-去保护”四步循环(图1.1),虽然只有“活化”与“氨解”这两步是用来形成肽键的,但氨基的“保护”与“去保护”又必不可少。导致多肽合成步骤冗长,操作繁琐,原子经济性低。为了形成一个酰胺键,上百个原子都被浪费掉了。这种情况在固相多肽合成(SPPS)中更为严重,由于固相多肽合成是非均相反应,为了保证产品的纯度和总收率,反应所需的缩合剂、添加剂和原料氨基酸都是大大过量的,每生产一公斤肽就会产生数吨的化学废物。因此,传统多肽化学合成面临着绿色化学的严峻挑战,“高原子经济性”的绿色酰胺键(或肽键)构建方法在过去二十年内被两次遴选为“有机合成化学领域的十大挑战”之一。
随着越来越多的多肽药物上市(尤其是生物利用度相对较低的口服多肽药物),对多肽原料药的需求急剧增长,迫切需要符合可持续发展理念的绿色多肽合成方法。多肽合成绿色化成为多肽合成领域的重要研究前沿,科学家在绿色溶剂、液相载体、流动化学及纳滤技术等层面对多肽合成绿色化进行了深入探索并取得了一些进展,但是这些工作大多集中在技术层面,未涉及肽键形成原理上的创新。从肽键形成原理上来讲,保护基的使用是造成多肽合成原子经济性低的根本原因。如能直接以无保护氨基酸来合成多肽,将省去“保护”与“去保护”步骤,极大地提高多肽合成的步骤经济性和原子经济性。其实科学家早在60多年前就提出了无保护氨基酸合成多肽的设想,一种策略是将氨基酸的保护与活化融为一步,利用五元环状活性中间体与正在增长的肽链的N端氨基形成肽键并释放出新的氨基用于下一个氨基酸的偶联,实现肽链从C端向N端的延伸,但这个新产生的氨基会与当前的活性中间体继续反应,形成聚合副产物(图1.2)。为了避免C→N多肽合成过程中的聚合副反应,在核糖体多肽合成原理的启发下,科学家提出了无保护氨基酸N→C逆向合成多肽的策略。但逆向多肽合成过程中严重的消旋问题导致该策略一直未能成功。这是因为逆向多肽合成涉及多肽片段C端羧酸的反复活化与氨解,而多肽C端羧酸α-手性中心的外消旋化速度通常是烷氧羰基保护氨基酸的35–110倍,迫切需要发展新型的不消旋缩合试剂和缩合方法。
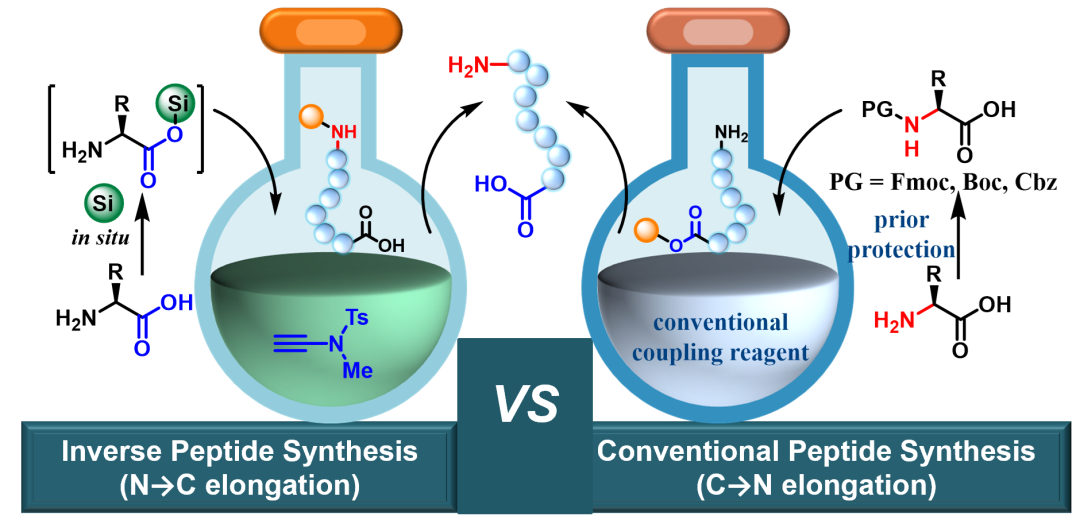
赵军锋教授课题组一直致力于通过发展有机化学新反应和新试剂来解决多肽化学合成领域的关键科学问题。他提出了以稳定烯基活化酯为中间体的缩合试剂设计概念,发现了炔酰胺(J. Am. Chem. Soc. 2016, 138, 13135-13138)与联烯酮(J. Am. Chem. Soc., 2021, 143, 10374-10381)两类原创型多肽缩合试剂,由于其促成酰胺键形成时的作用机制与传统缩合试剂完全不同,这两类缩合试剂在抑制氨基酸活化与氨解过程中的消旋时展现出了显著的优越性,为解决多肽合成过程中α-氨基酸手性中心的外消旋化问题提供了一种强有力的工具。因其可有效避免用于多肽片段羧酸活化与氨解时的消旋,炔酰胺类缩合试剂不但能够用于简单酰胺和二肽的合成,还可用于多肽片段连接和首尾环化 (Angew. Chem. Int. Ed. 2022, 61, e202212247;Green Chem. 2021, 23, 9916-9921; ACS Catal. 2020, 10, 5230-5235.) 。使肽链沿N→C逆向无消旋延伸成为可能。最近,赵军锋教授课题组利用炔酰胺为缩合试剂,通过现场保护的策略,间接实现了以无保护氨基酸为原料的逆向多肽合成,破解了困扰多肽合成领域60多年的难题。相关成果发表在化学领域国际顶级期刊《美国化学会志》(J. Am. Chem. Soc.)上,广州医科大学药学院在读博士研究生刘涛同学和已毕业硕士研究生彭泽军同学为该论文的共同第一作者,赵军锋教授为唯一通讯联系人,在读硕士研究生赖曼婷同学和胡隆副教授也做出了重要贡献。
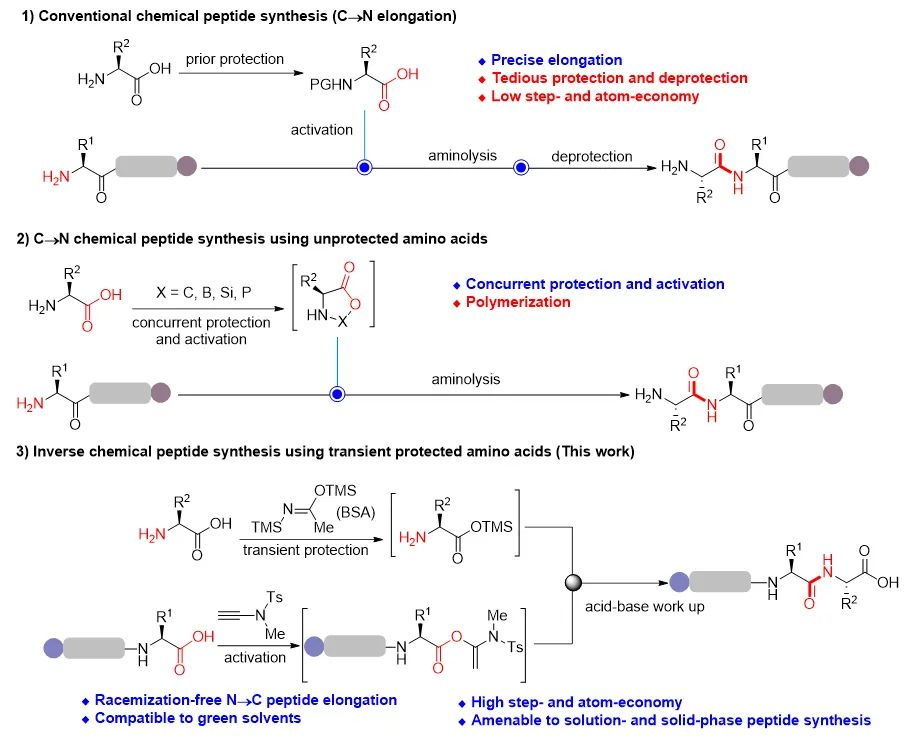
图1. 1)以氨基预保护氨基酸为砌块的传统C→N多肽合成;2)以无保护氨基酸为原料的C→N多肽合成;3)以瞬态保护氨基酸为砌块的N→C逆向多肽合成
由于无保护氨基酸是以两性离子的形式存在,不但溶解性差,而且亲核性弱。他们利用硅试剂可将两性离子型无保护氨基酸的羧基现场自发转化为硅酯的特性,发展了一种瞬态保护策略。硅酯的形成不但解决了溶解性问题,同时释放出具有强亲核性的氨基,与另一分子羧基活化的氨基酸形成肽键。更重要的是硅酯对水、质子性溶剂和弱酸都非常敏感,极易发生水解,在肽键形成后仅需简单的酸碱处理即可脱硅释放出羧酸官能团。他们用炔酰胺缩合试剂将第一个氨基保护的氨基酸的羧基现场定量转化为α-酰氧基烯酰胺进行活化,然后与瞬态保护的第二个氨基酸硅酯的氨基进行缩合,肽键形成后通过酸碱处理即可得到N端保护的二肽羧酸。不像其他缩合试剂在用于该策略时仅限于二肽羧酸的合成,炔酰胺缩合试剂还能够用于三个氨基酸以上的肽链的高效合成,先用炔酰胺缩合试剂对二肽羧酸进行活化,再与第三个瞬态保护的氨基酸硅酯的氨基进行缩合即可得到三肽。由于硅酯可在酸碱处理过程中自发释放羧基,因此无需额外的脱保护步骤来去除瞬态硅酯保护基,从而实现瞬态保护、活化、氨解和原位脱保护在一锅中进行,反复重复这一循环即可获得目标多肽。他们利用无保护氨基酸为原料成功合成了一系列含5-9个氨基酸的多肽原料药(图2)。由于瞬态保护、活化、氨解和原位脱保护均在一锅内进行(可认为是一步操作),与传统多肽合成方法相比,该策略大幅缩短了反应步骤。以含有8个酰胺键的赖氨加压素(Lypressin)为例,传统方法需要24步才能构建这8个肽键,而该策略仅需8步操作即可实现。该策略底物范围广,20种天然α-氨基酸(除组氨酸外)无论是作为羧酸组分还是氨基组分都能以几乎定量的收率构建肽键。同时该策略对羟基、巯基、伯酰胺、吲哚、咪唑等氨基酸侧链官能团都有很好的兼容性。β-氨基酸、γ-氨基酸和N-甲基氨基酸等非天然氨基酸也同样能以优秀的收率得到目标产物。值得注意的是,大部分底物在反应结束后只需简单的酸碱处理和重结晶即可得到纯的目标多肽。
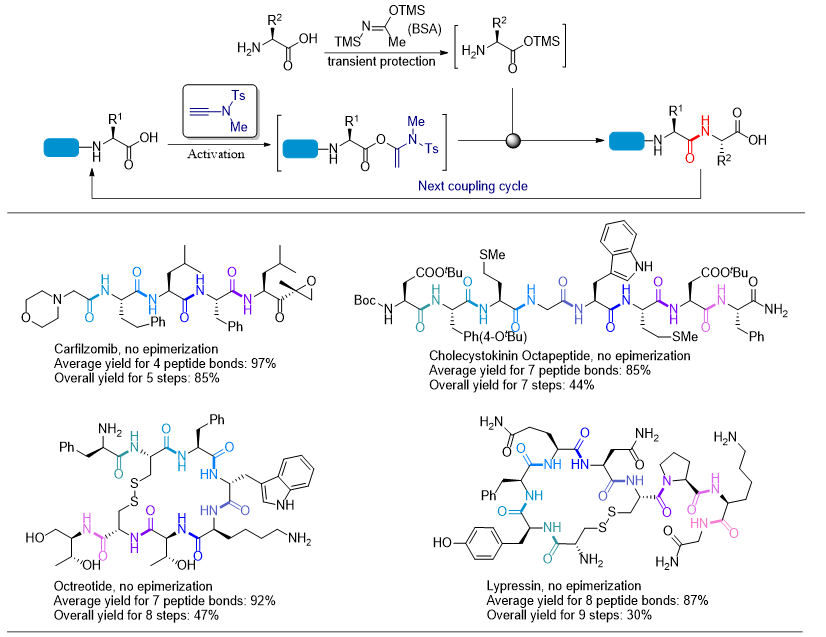
图2. 瞬态保护氨基酸逆向合成多肽原料药
该策略不但可用于液相多肽合成,而且可兼容片段连接和固相多肽合成。基于此瞬态保护策略,他们发展了间接以无保护氨基酸为原料的逆向固相多肽合成,并成功用于含21个氨基酸肽链的合成(图3)。因为多肽合成的常用溶剂N,N-二甲基甲酰胺(DMF)在欧洲已被禁用。他们还对绿色溶剂的兼容性进行了探索,发现该策略对碳酸二甲酯、碳酸二乙酯、2-甲基四氢呋喃等绿色溶剂很好的兼容性,为其规模化应用奠定了基础。
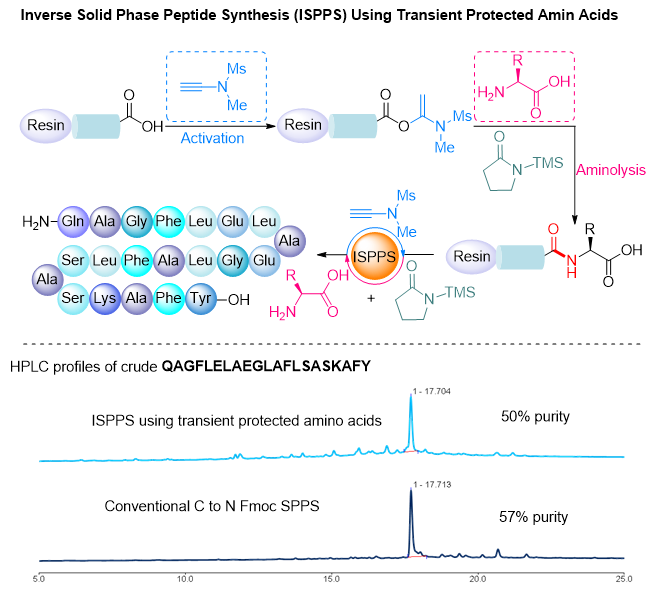
图3. 瞬态保护氨基酸在固相载体上逆向合成多肽
总结
赵军锋教授课题组利用他们原创的炔酰胺缩合试剂,通过瞬态保护的策略,间接实现了以无保护氨基酸为原料的逆向多肽合成。该工作被Science杂志新闻评论员Derek Lowe博士以“A New Way to Synthesize Peptide”为题作为科研新闻进行了报道 [1],称赞该策略大幅缩短了反应步骤,减少了纯化操作,显著提高了多肽合成的步骤经济性与原子经济性。其反应条件温和,操作简单,能够兼容绿色溶剂,原料氨基酸价格低廉,该策略不但可用于液相多肽合成,还可用于固相多肽合成和片段连接,有望大幅度降低多肽合成的成本,提升多肽药物的可及性。相信该工作将激发更多的理论和技术创新,推动多肽合成跨入无保护氨基酸时代。











