
当前位置: 行业资讯
Penicibilaenes A和B的首次不对称全合成
本文转自X-MOL
三环倍半萜是一类重要的天然产物,它们的结构多样性和生物活性不仅引起了人们对药物发现领域的兴趣,也是化学家发展创新方法和策略的灵感来源。三环[6.3.1.01,5]十二烷骨架是[3.3.1]-桥环和[4.3.0]-并环组成的结构单元,广泛存在于众多具有重要生物活性的复杂天然产物和药物分子中,关于该环系骨架的构建吸引了大批有机合成化学家的关注。
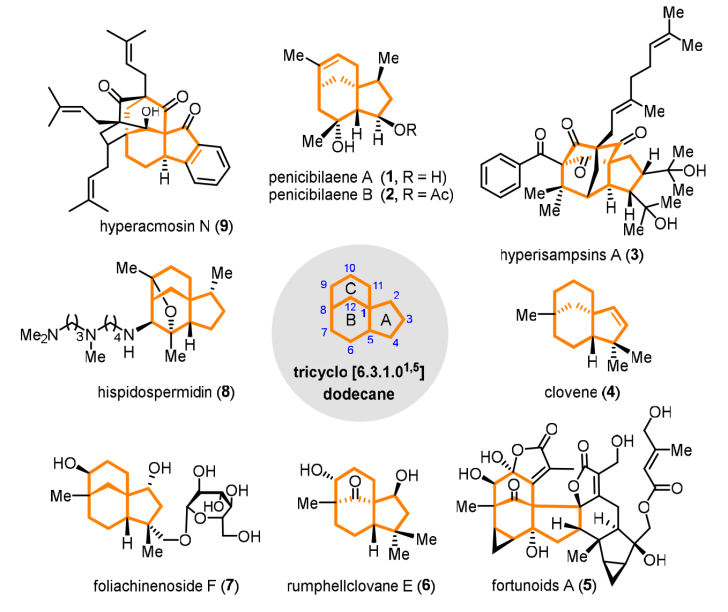
图1. 具有[6.3.1.01,5]十二烷骨架结构单元的代表性活性天然产物
天然产物(+)-penicibilaenes A和B是中国科学院海洋研究所王斌贵教授课题组于2014年从海洋青霉属真菌Penicillium bilaiae MA-267中分离得到的具有全新三环[6.3.1.01,5]结构的倍半萜天然产物。该天然产物具有抗真菌活性,对植物病原真菌Colletotrichum gloeosporioides表现出选择性抑制活性,特别是penicibilaene B的抑制效果优于广谱抗生素zeocin。2021年,董广斌教授课题组首次利用“C-C键活化/C-H键官能团化”策略成功实现了penicibilaenes A(1) 和B(2) 的首次外消旋体全合成。同年,Kazuyuki Sugita教授课题组以分子内Aldol 缩合和RCM反应为关键反应,完成了penicibilaenes A(1) 和B(2) 的外消旋体全合成。
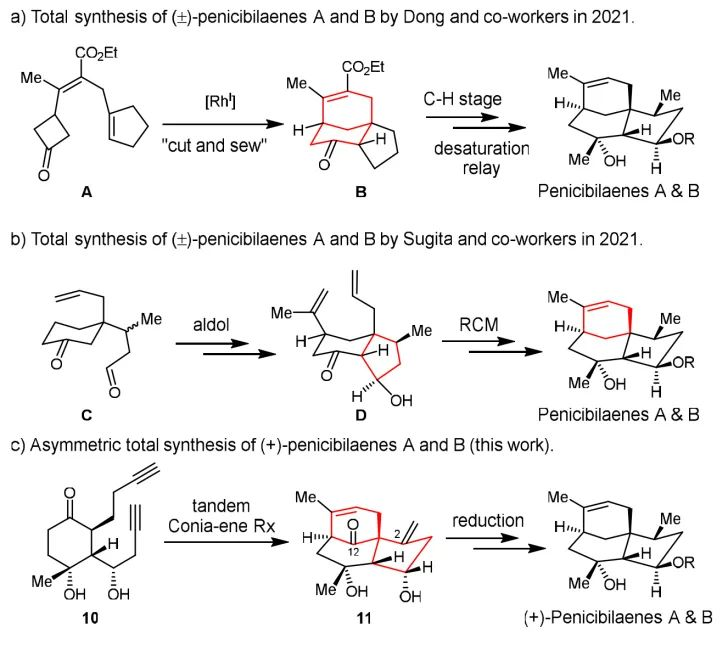
图2. penicibilaenes A和B的全合成策略分析
近日,杨震教授课题组发展新的合成策略,实现了(+)-penicibilaenes A和B的首次不对称全合成。该合成路线主要有以下特征:1)手性辅基诱导的1,4-共轭加成;2)非对映选择性的1,2加成构建C4,C6手性中心;3)tBuOK/DMSO反应体系促进的Conia-ene型串联环化反应;4)C4位羟基导向非对映选择性构建C2手性中心。

图3. penicibilaenes A和B的逆合成分析
首先,作者从合成手性二炔基化合物10开始。如图4所示,从商业可得的1,4-环己二酮出发,经过两步已知反应得到烯酮16。以(R,R)-hydrobenzoin为手性辅基的烯酮16可以诱导立体选择性的铜催化1,4-共轭加成反应,然后与HC(OMe)3发生原位甲酰化反应,以58%的产率得到共轭加成产物18和19,其中非对映选择性比例为1:1.8(主要产物19为所需目标化合物,同时化合物18经衍生化也以单晶形式得以确认其立体化学)。之后经三氟乙酸(TFA)脱除化合物19中的缩醛保护,得到的醛基化合物15与炔丙基格氏试剂21在催化量ZnBr2的作用下,以单一非对映选择性得到目标产物22,收率为72%。高非对映选择性可能来源于形成了15a中的Mg2+螯合中间体,使得亲核试剂仅能从si面进攻。为了引入23中C6立体手性中心,作者通过添加镧系金属盐LaCl3•LiCl,与MeMgCl和底物在0°C下反应得到二醇23,产率为60%。22a中形成的Mg2+螯合中间体同样对该反应的非对映选择性发挥了关键作用。之后经脱保护以56%的产率得到关键反应前体二炔基化合物10。
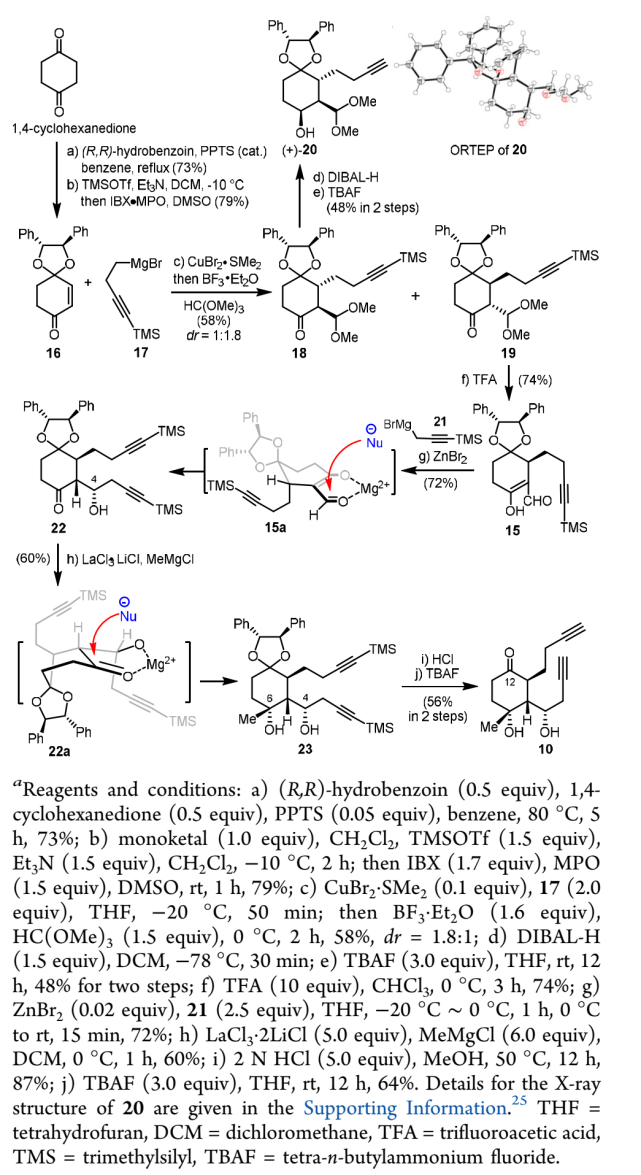
图4. 二炔基化合物10的不对称合成
随后,作者开始探索关键的Conia-ene型串联环化反应,构建核心三环[6.3.1.01,5]十二烷骨架。2017年,Dirk Trauner教授课题组报道了tBuOK/DMSO反应体系促进的分子内Conia-ene型反应,用于构建含五元环的结构单元。以二炔基化合物10为底物,在0.5当量的tBuOK反应条件下,以34%的产率得到目标产物11,作者并未观察到单次环化的产物14,延长反应时间也并不能提高反应产率。在5.0当量的tBuOK反应条件下,该反应产率提升至64%,且反应原料在10分钟内转化完全。在此,对于目标产物11中C1和C8的立体选择性,作者推测可能归因于二炔化合物10在串联环化反应过程中所呈现的有利构象,使所得到的环己酮烯醇化物从re面与炔键反应。
接下来,作者将注意力转向(+)-penicibilaenes A (1) 和 B (2)的最终全合成研究。在C2位立体手性中心的构建中,作者采用了区域和非对映选择性氢化的合成策略。首先,作者尝试了几种经典的过渡金属催化氢化反应条件和氢原子转移(HAT)反应条件,但均未能得到理想结果。之后,在Crabtree催化剂的反应条件下,利用C4位羟基导向非对映选择性还原C2位环外双键,以78%的产率得到目标产物12。化合物12经Wolff -Kishner-Huang还原得到化合物25,之后经C4羟基官能团的氧化/还原反应,翻转羟基手性中心,以2步56%的产率得到penicibilaene A (1)。从1出发,经C4位羟基选择性乙酰化反应,得到天然产物penicibilaene B (2)。
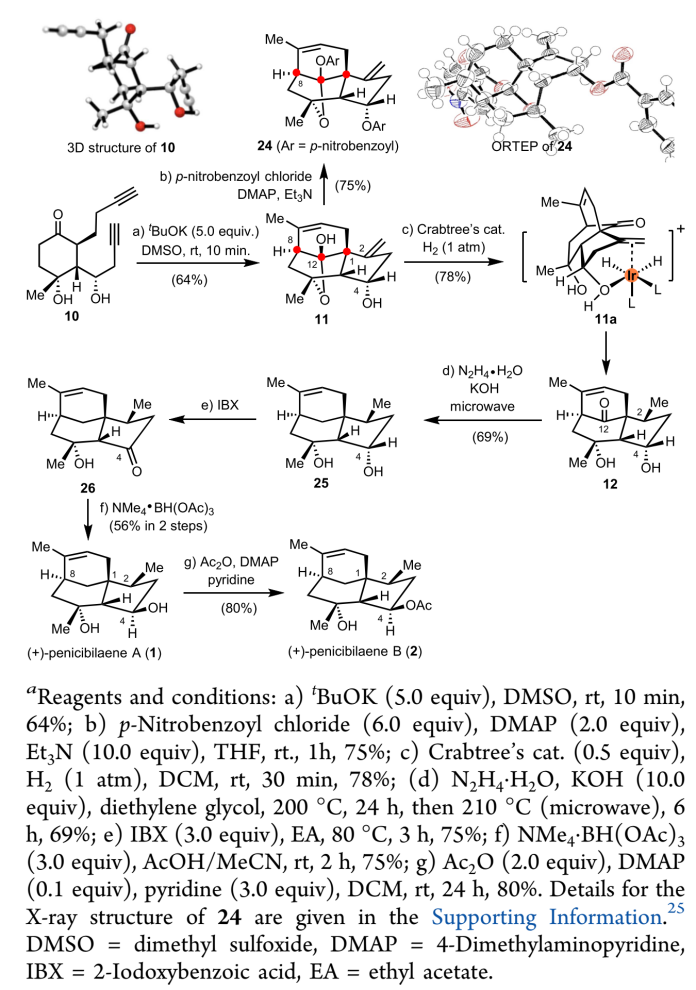
图5. (+)-penicibilaenes A和B的全合成
之后,作者对关键的Conia-ene型串联环化反应的可能反应机理进行系统研究。对于化合物11中通过该关键反应形成刚性三环[6.3.1.01,5]十二烷核心骨架的意外结果,引发了作者的思考:1) 考虑到该反应较快的反应速率,其中是否涉及自由基类型的反应机理? 2)为什么没有分离得到串联反应中间体14? 3) C12上的羰基是否参与了C9-C15位双键的异构化过程? 为了回答这些问题,作者进行了一系列的实验。首先在标准反应条件下,添加过量的自由基捕获剂TEMPO,该关键反应的反应速率和产率并未受明显影响,表明该反应不是通过自由基反应机制。接下来,作者通过DFT理论计算来解释反应历程。通过比较二炔基化合物10在5-exo-dig和6-exo-dig环化时的反应能垒(ΔΔG‡=5.9 kcal/mol),说明优先进行5-exo-dig环化得到产物13。并且中间体14经过过渡态TS14得到中间体13的反应能垒较低(17 kal/mol),可能是导致中间体14在室温下未分离得到的原因。随后作者分析了C12羰基是否参与了从中间体13到目标产物11的双键异构化过程。在中间体13去质子化过程中,从反应吉布斯自由能比较来分析,攫取C10氢的过渡态TS13b比攫取羰基α位C8氢的TS13a的吉布斯自由能低7.2 kcal/mol。如图6所示的Newman投影式,攫氢过程中的二面角分别为28°和99.4°,说明TS13b能通过有效的CH−π轨道重叠具有更强的稳定性,从而更加有利于去质子化过程的进行。
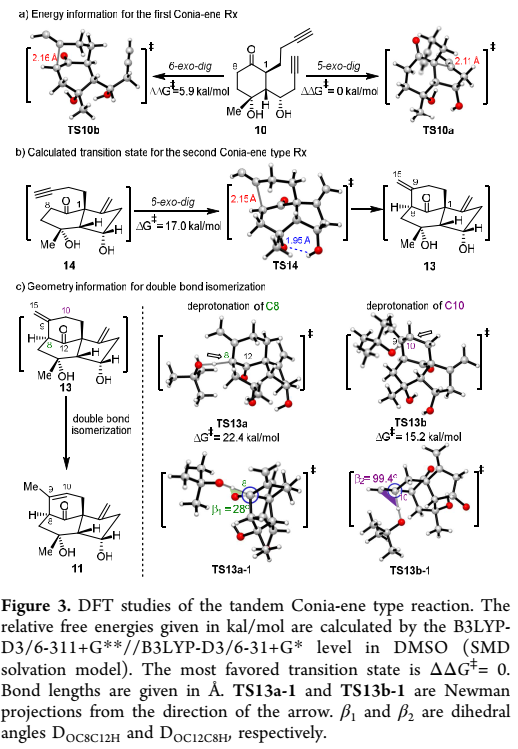
图6. 关键的Conia-ene型串联环化反应机理研究
总结
杨震教授课题组由市售原料1,4-环己二酮出发,分别经14步和15步转化,完成天然产物(+)-penicibilaenes A(1) 和B(2) 的首次不对称全合成研究。其关键合成策略为tBuOK/DMSO介导的Conia-ene型串联环化反应构建刚性三环[6.3.1.01,5]十二烷骨架,同时该合成策略也为其他具有三环[6.3.1.01,5]十二烷核心骨架结构的活性复杂天然产物的高效、简洁全合成研究提供了一条有效的途径。
该成果的第一作者为北京大学深圳研究生院博士研究生王哲源,博士研究生宋治霖对DFT理论计算做出了重要贡献,南华大学黄俊教授/北京大学深圳研究生院杨震教授为通讯联系人。该工作得到了国家自然科学基金、青岛海洋科学与技术国家实验室等项目的资助。











